

Imaging-Based DNA Sequencing: Past, Present & Future [Part One]
How image analysis and computational tools have taken genome sequencing from years to minutes; from population to personal
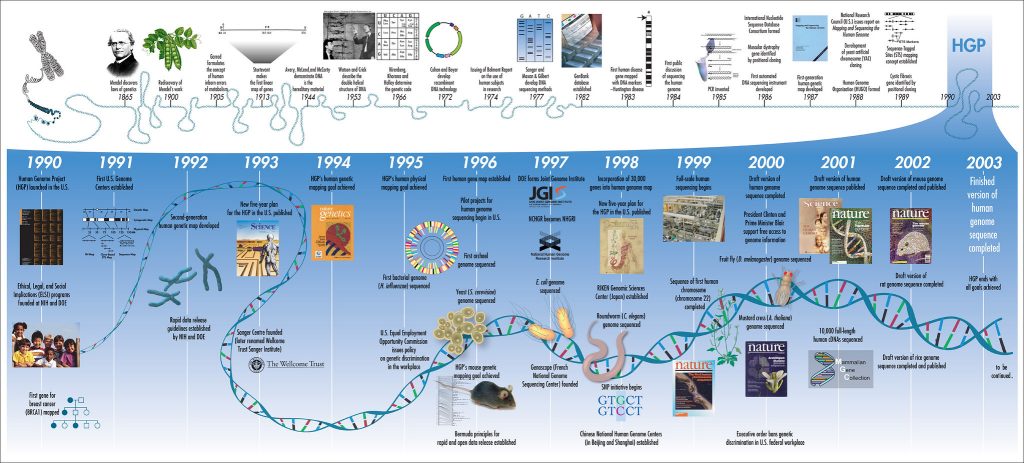
DNA sequencing has improved exponentially in recent years. The Human Genome Project took 13 years and nearly $3 billion USD to sequence the entire human genome, finishing in 2003. And even this was a rough draft and prone to errors. Now there are sequencing techniques that can be completed in real time, where entire genomes are sequenced in minutes or hours and the cost is as low as $1000 USD per genome. Today the market is growing rapidly thanks to the declining cost of sequencing procedures, new government funding, the rise of consumer solutions, and the increasing prevalence of chronic diseases. Between 2020 and 2025, the market is set to double to 50 billion dollars.
As with all scientific advancements, there are a multitude of drivers behind it. Dramatic changes in image analysis and computational tools have been essential in making these leaps. For example, data storage alone has made a significant contribution. Just 15 years ago a typical USB thumb drive was less than a gigabyte, and cloud storage wasn’t widely available. The data from a single genome could take up 200 gigabytes. For a lab that utilizes imaging data, each researcher generates terabytes of images every year, so fast and cheap storage is a must.
Early Genomics: Creating A New Science
Compared to modern methods, the original Sanger Sequencing process is arduous and material-heavy. It relies on dideoxynucleoside chain-terminating reactions, where each of the four DNA nucleotide building blocks are labeled with a radioisotope marker. These four reactions are then run on a polyacrylamide gel in four separate lanes, where the various fragment lengths are separated out by the electrical field. X-ray imaging is used to detect the radiolabelled nucleotides, thus identifying the base pairs and their location, which is used to build the sequence.
The Sanger method is a slow and manual process, in it’s original form it could take several days to sequence just 50 base pairs [Sanger & Coulson 1975]. In addition to being slow and labor-intensive, the original Sanger method is prone to error, but it does have the advantage of utilizing fairly minimal data storage and basic imaging techniques. Even from these early days it was clear that computer-aided methods would be required to automate the processing of data. As researchers sequenced more and more genomes and built larger reference libraries, they increasingly needed tools that could compare the new data with existing sequences to find overlap. It was around this time that the NIH founded GenBank to store sequence information. Genbank allows researchers to access an annotated collection of publicly-available sequence data for over 300,000 organisms. This incredible, collaborative tool allows labs to easily compare new sequences with previously decoded ones and find overlap, evolutionary conservation, or even pick a target sequence for mutation. GenBank started with 680,000 base pairs and 606 sequences in 1982; by 1992 this increased nearly 200x, and nearly 42,000x by 2002! This exponential growth continues, thanks to faster, more accurate methods coming online.

The initial use of the Sanger method was incredible and exciting, but many labs were creating faster ways to implement it. One such creation was facilitated by the discovery and manipulation of the Green Fluorescent Protein (GFP). Smith & Hood produced fluorescently labeled DNA primers in 1985, which they used in conjunction with Fluorescence Energy Resonance Transfer (FRET) to sequence in a semi-automated fashion. In 1987, Prober et al. demonstrated that four different fluorophores tagged to chain terminating dideoxynucleotides could be used to map a sequence using a single vial for sample preparation and a single lane for gel electrophoresis, instead of the four vials and lanes in the original Sanger method.
Directly imaging the gels to determine the fluorophore for each fragment size meant a faster, more precise building of the DNA sequence. However, the gel run still took 6 hours in this new method, with additional time needed to analyze the data produced. This updated method allowed researchers to sequence up to 600 bases an hour, which was a huge advancement over the original Sanger method. But the human genome is roughly 3 billion bp, so at that rate it would have taken over 500 years to sequence it.
In 1986, Applied Biosystems brought the ABI 370A automated sequencer to market, which could process up to 32 samples per run, more than doubling the methods by Prober and team. This system relied on early computer algorithms to correctly identify the base pairs and order, which would become more and more critical for improving output.
When the Human Genome Project was proposed in 1990, they estimated it would take 15 years. This estimate was very optimistic if they were only using the technology available when they started. But rapid improvements were on the horizon.
One area for improvement was imaging, specifically through improvements in fluorescent labels. To understand the critical improvements made by developing better fluorescent tags and dyes, it is important to understand the dominant imaging technique for sequencing, FRET. FRET is what allows all four fluorescent tags to be excited by a single wavelength– but emit different signals that the detector /camera can distinguish. Typically in fluorescence imaging, you will use one fluorophore for a given experiment, then excite that fluorophore (the light from a laser [photon] energizes the electrons of the fluorophore) and you can capture the corresponding emission (the release of a photon from the fluorophore – energy is lost in the transition, so the photon released is lower energy and emits a longer wavelength of light) to create an image. You might even use multiple fluorophores, but each would need to be excited by a different wavelength supplied by a separate laser to be able to distinguish them from one another (e.g. a 488nm laser will excite GFP, a 591nm laser will excite Red Fluorescent Protein, or RFP). FRET allows scientists to image multiple fluorophores, which emit at very different wavelengths (sufficient spectral overlap to allow them to be distinguished from one another) using a single excitation laser. This works by a donor fluorophore absorbing energy in the form of a photon, then transmitting that energy to an acceptor fluorophore (not in the form of a photon), and the acceptor then releasing that energy as a photon.
The same donor fluorophore can be used for all four nucleotides, so one excitation laser can be used – but emission of four different wavelengths based on the base pair can be achieved by using four different acceptor fluorophores. This, combined with computational methods, allows for real-time imaging and base-pair calling, substantially increasing the speed of sequencing, as demonstrated by Applied Biosystems in the late 80s. Further improvements to this technique were made through the advancement of fluorescent labels. New dyes developed specifically for energy transfer were able to improve signal-to-noise ratios by 4-5 fold, making base calling more accurate.
In addition to improving fluorophores and image analysis, researchers were also focused on creating longer sequence read lengths. The Sanger method created read lengths of ~800bp, but the full sequence for even simple organisms are thousands to millions of base pairs, and the human genome registers in at billions of base pairs. So, researchers needed a way to sequence longer segments. They began using “shotgun” sequencing, where they fragment the DNA, clone it to high copy numbers using the newly invented Polymerase Chain Reaction (PCR), and then sequence the fragments. Computational models are used to align the fragments into a contiguous sequence based on overlapping sequences. This allowed for faster assembly of longer sequence segments. This technique combined with capillary electrophoresis, which provided faster run times and better separation of the fragments, and in line FRET imaging is what is known as semi-automated Sanger Sequencing. Nearly 1000bp read lengths at 99.999% accuracy were achieved this way.
Applied Biosystems remained the market leader throughout the ’90s, improving and released new technology each year including machines that used capillary electrophoresis and fluorescence imaging to create massively parallel experiments. By 1998, 1000 bp could be sequenced in less than an hour – but there were new techniques and technologies still on their way.
Next Generation/Second Generation Sequencing – Better Accuracy and Speed
The early 2000s were marked by a significant shift in the market as many new players entered. Next Generation Sequencing (NGS) technology was a key new method, which used many cycles of amplification to create huge amounts of DNA for sequencing. The combination of much larger amounts of DNA and the miniaturization of the sequencing process using microfluidic devices means that hundreds of millions of sequences can be produced in parallel. This produced much faster, more accurate sequencing results – though often with significantly shorter read lengths (35-500 bp, vs the 800 bp by Sanger sequencing). Computational tools were developed to overcome the short read lengths and sequencing costs plummeted. 454, Solexa, Illumina, Ion Torrent and others joined Applied Biosystems in the development of NGS technology.
In particular, the high throughput technique for pyrosequencing was introduced, which utilized the light generated through pyrophosphate synthesis instead of fluorophores. This technology was commercialized by 454 Life Sciences in 2003 with the GS20. This machine could sequence up to 25 million base pairs in a single 4-hour run, enabled by CCD sensors beneath nearly 1 million micro-wells where the sequencing reactions took place. This was a disruptive technology, using an entirely different method, and could sequence DNA lengths of ~500 bp with 99% accuracy.

Soon after, Solexa was founded, using sequencing by synthesis techniques that relied on fluorescent labels; essentially the Sanger method but at a much larger scale. This change in scale was facilitated by miniaturization, the use of microfluidics and improvements in process automation. Acquisition of colony sequencing (bridge amplification) technology from Manteia allowed for stronger fluorescent signals, and therefore improved base calling. They also developed an engineered DNA polymerase and reversible terminators – which led to a platform that could read individual base pairs as they were added to the sequence. This platform produced up to 1M bp per run, but sequence lengths were relatively short, at only ~35bp. In 2007 Illumina acquired Solexa and set out to become a market leader in NGS.
In part two, we will explore the current state of image-based genomic sequencing and what we can expect in the future.
 Advancing genome sequencing at the speed of light
Advancing genome sequencing at the speed of light  Lock And Load: Shotgun Metagenomics and SARS-CoV-2
Lock And Load: Shotgun Metagenomics and SARS-CoV-2 