

Genomics takes its place in the race against COVID-19
Using genomic technology on the trail of the latest coronavirus – where it came from and where it’s going next
Advancements in genetic technology are making it faster, easier, and less expensive for public health experts to understand how this new coronavirus spreads. Time is of the essence for the people on the front lines of this outbreak as the virus has already sickened tens of thousands.
What is COVID-19?
Named for its unique shape (from Latin “corona,” meaning “crown” or “wreath”), the coronavirus represents an entire category of viruses that are thousands of years old. While most common in warm-blooded flying vertebrates like bats and birds (its ideal hosts), its ever-evolving variants can infect other animals, like cattle, canines, and humans.
In humans, coronaviruses cause respiratory tract infections that can vary, from a mild case, like a common cold, to other infections that can be lethal, like SARS (2003), and MERS (2012). Now there’s COVID-19, the name of the disease caused by a new coronavirus named Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2), identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in late 2019.
While epidemic and pandemic diseases are often compared to the annual global flu outbreak, it’s COVID-19’s newness that makes it a unique challenge. Public health officials and governments know the risks of the flu. They can already predict its patterns and impact, allowing them to manage its effects (through both educational and medical measures).
Experts still don’t know how dangerous the new coronavirus may be, and while it’s still not widespread, there’s still a chance to stop it. That was the case with SARS, which through global coordination, was contained in 2004. But there was never a vaccine developed. There are yet to be vaccines or antiviral drugs to prevent or treat any human coronavirus infections. Scientists started work on drugs to treat coronaviruses during both the SARS and MERS outbreaks, but because the outbreaks died down, the job was never completed.
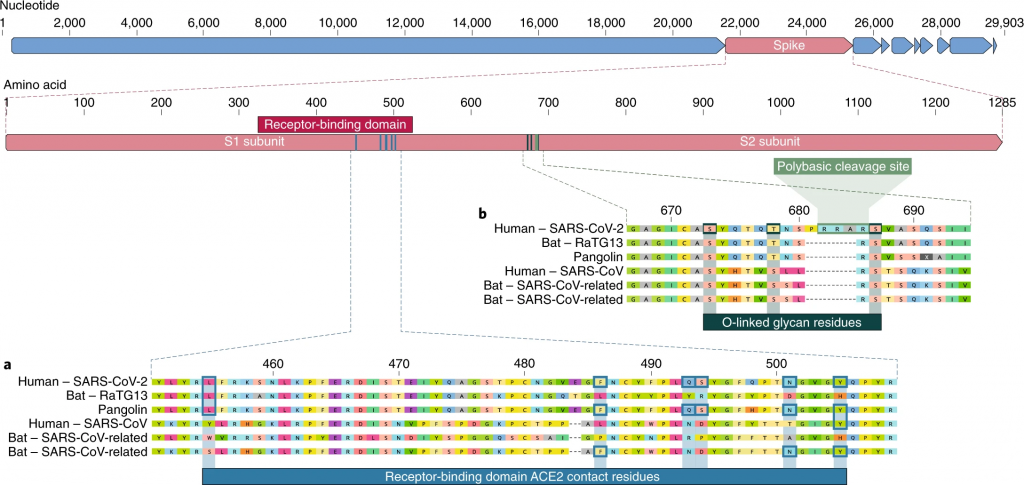
In 2002, when scientists sequenced the 29,751-base genome of the coronavirus related to SARS, they found that it was not closely related to other known coronaviruses, including two other human coronaviruses (HCoV-OC43 and HCoV-229E). Knowledge of the complete genome sequences of SARS-CoV-2 is crucial to the diagnosis of coronavirus infections in humans and potential animal hosts, in the development of antivirals, and will ultimately facilitate the design of drugs and vaccines to cure or prevent it.
Where did SARS-CoV-2 come from?
Zeng-Li Shi , lead investigator at the Center for Emerging Infectious Diseases at the Wuhan Institute for Virology, has been studying coronaviruses for several years, particularly in bats. Her work has shown that the southern, subtropical areas of Guangdong, Guangxi and Yunnan have the greatest risk of coronaviruses jumping from animals to humans. After years of sampling, Zeng-Li and her team of researchers found a diverse group of coronaviruses, hundreds even, in multiple bat species. In many cases, multiple viral strains could infect each bat. Spillover into other species is apparently common.
Why bats? Recent research has shown that bats have a rather fierce immune response to viruses, driving viruses to replicate and adapt faster to survive. When a virus finally makes the jump to mammals with more typical immune systems, such as humans, the adapted viruses wreak havoc. “Bats” is also an enormous category. There are over 1,250 bat species in existence, equal to about one fifth of all mammal species. That’s a lot of (genetic) variety. In these lush areas, filled with fruit and nut trees as well as caves, it’s hard to avoid bats. They roost in homes and eat crops, and are disturbed by people exploring caves, mining guano, or even hunting them. Zeng-li has also found that people who didn’t have any direct contact with bats could still exhibit antibodies or symptoms of infections, possibly from respiration or fluid droplet.

So innovative virus research in a populated area is one factor. But in the more serious cases, it seems an intermediate step between bat and human is necessary. Genomic research on a version of SARS-CoV found in civets (small, lean, mostly nocturnal mammals native to tropical Asia and Africa) revealed that this version was a 99.8% match with the human one, much closer than the one found in bats, indicating that’s where the virus jumped first. With MERS, camels were an intermediate host before the virus jumped to humans.
The civet version of SARS-CoV is much more closely related to ours than the bat-to-human varieties of SARS-CoV-2. Researchers are now looking for this intermediary, and evidence suggests that pangolins are a strong potential candidate. This same genomic research was useful in eliminating another potential source – that the virus itself was man-made. You may or may not be comforted by the idea that the ‘spikes’ on SARS-CoV-2 are actually not well-optimized to infect humans – we are apparently capable of designing something much more effective.

Detailed understanding of how an animal coronavirus jumped from different species to infect humans so successfully will help prevention in the future. For example, if SARS-CoV-2 adapted in another animal species and infected us, it could easily happen again. But if the virus adapted in humans alone, then it’s unlikely that this same series of mutations would happen twice.
Understanding the genome of SARS-CoV-2
Researchers got an early win in January. It took only two weeks after public health officials reported the virus to the World Health Organization for Chinese scientists to isolate the virus and figure out the full sequence of its genetic material. This sequencing answered the most basic question about the disease: What pathogen is causing it? The genomes that followed were almost identical, suggesting the virus, which originated in an animal, had crossed into the human population just once. If it had jumped the species barrier multiple times, the first human cases would show more variety. This gave us a sense of what we were dealing with, but also ideas of how to track and treat it.
Since then, the virus has been isolated and sequenced by China, Japan, Thailand, the US, India, and Canada. As soon as that sequence was public, biotechnology companies started creating synthetic copies of the virus that could be used in research. Testing kits are being developed around the world. It also gave scientists the ability to start creating vaccines. Dozens are under development at pharmaceutical companies.
This is all happening faster than ever before. During the SARS outbreak in 2002, it took months to sequence the virus’ genome, and even longer to replicate it in the lab. This has been possible because next-generation sequencing (NGS) technologies can quickly capture the virus from patient samples and generate whole genome sequences. This will help medical experts to characterize the virus more quickly. Their work may help to track the evolution of the virus and trace its spread.
Genomic sequencing and synthesis is also much cheaper than it was two decades ago. In 2002 it cost nearly $100M to sequence the human genome. Today it costs less than $1,000. For synthesis, it used to cost $10 to create a synthetic copy of one single nucleotide, the building block of genetic material. Now, it’s under 10 cents. The new coronavirus gene is around 30,000 nucleotides long, so that reduction in price makes a big difference in how many copies scientists can make.
Coronaviruses: A moving target
The coronavirus is an RNA-based (ribonucleic acid) virus. RNA is a simpler (and believed to be older), single-stranded molecule that stores genetic information, but is much less stable than the DNA in our genome. RNA viruses like coronavirus mutate at high rates because RNA-based genomes do not copy themselves faithfully. They accumulate mutations that can quickly kill it or make it stronger. This is likely what happened in 2019: a coronavirus made a change and jumped from animal to human in a way that gave it an advantage. Obtaining more related viral sequences from animal sources would be the most definitive way of revealing viral origins. For example, finding an intermediate SARS-CoV-2-like virus from other animals would help us understand how the current virus was able to become so successful.
It will also help us know where it’s going. In Washington State, the first infection site in the US, researchers were able to compare the genomes of virus infections to understand how long it had been around. In the same way, Berlin virologists could track an infection in Italy back to Germany, originating in China. Or not – another interpretation proposed that the three different mutations found in Italy represented three separate paths from China. It was a case study in the power and pitfalls of real-time analysis of viral genomes. And the need for more data.
As the virus has raced around the world, more than 350 genome sequences have been shared on GISAID, an online platform for tracking infections. More diversity is emerging. Over the length of its 30,000-base-pair genome, SARS-CoV-2 accumulates an average of about one to two mutations per month. Using these little changes, researchers draw up phylogenetic trees, much like family trees, make connections between cases, and gauge whether there might be undetected spread of the virus.
While genomic differences offer clues to how the virus is spreading and evolving, they only represent a tiny fraction of cases and show few telltale differences, which are easy to over interpret. More data is needed.
The race to develop treatments for COVID-19
Work on both vaccines and treatments is progressing rapidly, but there is still a long way to go. Because so much is new, researchers are starting essentially from scratch on vaccine development. Vaccines help prevent people from getting sick in the first place. Treatments, on the other hand, help people after they are already sick. Treatments are further along and might be available sooner.
One possible treatment for COVID-19 that has seen considerable press is chloroquine or the closely-related hydroxychloroquine, medications used for a variety of autoimmune and malarial diseases. Doctors in South Korea, China, and the US are actively investigating these treatments and sharing what they’ve learned. In this particular case, chloroquine seems to destabilize the replication of the virus. This could provide a significant chance for treatment to cause the COVID-19 to self-destruct, giving more time for health systems worldwide to increase capacity and equipment as well as allow time for the public release of a vaccine. But it could lead to a mutation that makes it stronger or more harmful to humans. The only way to know is to learn more about the virus’ genome: how it works and how it’s changing.
(Editor’s note: while this was all true at the time of publication, there has obviously been a lot more in the news on this subject. As medications, these medications have potential upsides and downsides. We ask that you please take the advice of medical professionals.)
Looking for the crown today and tomorrow
Today, fully sequenced viral genomes are rare. But they will be crucial to future treatments. Technology that allows us to quickly generate accurate genomic data – especially when the viral genome is a moving target – will improve diagnostics and help researchers better understand how the virus develops and what can lead to better medical outcomes.
The availability of the SARS virus genome sequence is important, allowing researchers to focus on the features that make COVID-19 unique from similar related viruses. This genome-based understanding will form an important part of a public health strategy to control the spread of this disease as well as help us prepare vaccines for the inevitable variations that will crop up in the future.
*Current bulk sequencing equipment needs ultra-high speed cameras to provide high throughput of data to the massive computing equipment attached to the system. Teledyne DALSA has the camera technology to capture the photoemission signal from the microfluidic substrate. In addition to the technology, the company has nearly 40 years of experience in this high end technology domain. On the consumable side, the substrates which are actually a microfluidic chip require a wide portfolio of microfabrication technologies. Through its semiconductor division, Teledyne DALSA has more than 20 years of experience in MEMS technologies which include microfluidic chips which are part of the BioMEMS products and applications. Their microfabrication technologies include deep etching to form the microfluidic channels and substrate bonding to enclose from the top of those channels. Also BioMEMS require very high micro-machining accuracy where critical dimensions or feature size needs to be controlled in the nanometer range. The technology roadmap in NGS (Next Generation Sequencing) includes a different DNA reading approach where the DNA is sensed electronically instead of through an optical signal. Teledyne DALSA offers an integrated solution where the electrical CMOS readout chip can be designed by its Imaging division and the DNA BioMEMS reading cell can be manufactured at the Semiconductor division. Teledyne DALSA overall offers a unified portfolio of technology to further accelerate further technology development in partnership with companies already present or looking to break into the genomic world.
 A Hyperspectral Sight on the Stain
A Hyperspectral Sight on the Stain  CMOS TDI: A Game Changer in High-Fidelity Imaging
CMOS TDI: A Game Changer in High-Fidelity Imaging 