

Lock And Load: Shotgun Metagenomics and SARS-CoV-2
How Imaging-Based Genomics is Helping Us to Identify and Track the Virus Behind the COVID-19 Pandemic
Science must be exhausting sometimes. While the hope is that you discover or invent something new, you may be just as likely to reveal how little we know. In 2002, researchers in California used a new method of genomic sequencing to directly sample 200 liters of seawater.
What did they find? More than 5,000 different viruses. Almost all of them were new.
Other researchers applied the same method to several other samples and found similar rates of undiscovered viruses. They were there all along, but we didn’t know quite how to look for them. The then-new method is known today as metagenomic shotgun sequencing. This approach differed from existing methods in a few crucial ways. Genomic sequencing typically involved cloning a given DNA sample to make it easier to analyze. Not only could this be more time-consuming, but not all kinds of DNA could be cloned, meaning they wouldn’t show up in analysis.
The shotgun approach instead lets you look directly at a sample, and instead of looking for one specific thing, you’re mapping all of the DNA in a sample and using advanced computational techniques to understand what you’ve found.
The shotgun sequencing workflow
Shotgun metagenomics is not a single method. It’s a combination of high-throughput sequencing technologies with a suite of computational pipelines. It starts with fragmenting the entire genomic sample into varying sizes from 20-kilo bases to 300-kilo bases and then sequencing the fragments.
One way to do this is with imaging, called “sequencing by synthesis”: fluorescently labeled bases are added to single DNA strands from a sample and read in parallel. The light emissions are recorded by specially-filtered CMOS sensors looking for evidence of one of the four possible base pairs. The process simultaneously identifies DNA bases while incorporating them into a nucleic acid chain. Each base emits a unique fluorescent signal as it is added to the growing strand, which is used to determine the order of the DNA sequence. This sequencing cycle is repeated “n” times to create a read length of “n” bases.
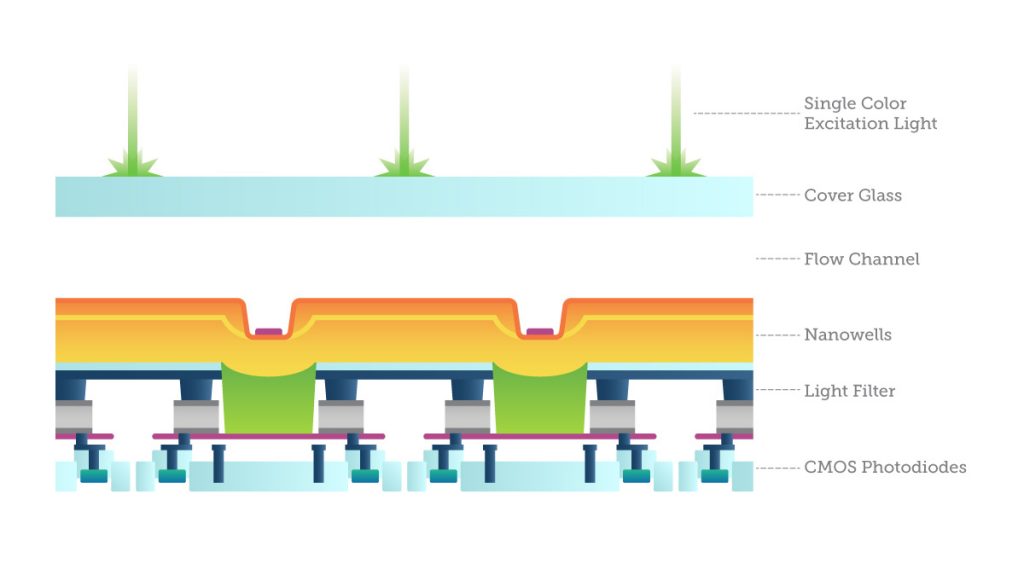
After sequencing, it is necessary to assemble the fragments by looking at the overlapping regions using sophisticated computer software. This is enormously computationally demanding. One researcher compared it to getting a DNA sample from every person living in New York, all at once, mixing it all together in small pieces, and then trying to figure out who was who.
Since existing genome maps are not used, as in other methods, there are more likely to be errors during the assemblage. Also, since shorter fragments are used, they may provide less unique information for each read, requiring more samples to work from.
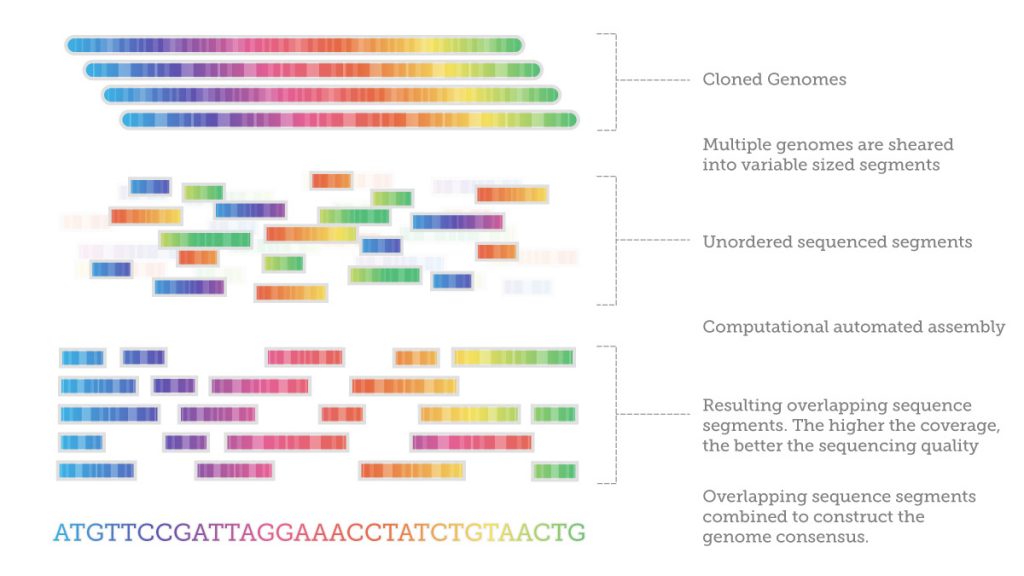
Or just smaller genomes: the shotgun sequencing method is currently the most efficient and cost-effective strategy for sequencing microbial genomes: bacteria, viruses, and yeast. Their smaller genomes lack repetitive regions which are difficult to sequence (by comparison, up to 50% of the human genome is repetitive), it’s possible to assemble these genomes more easily without errors.
Genomes like SARS-CoV-2.
Using shotgun metagenomics against COVID-19
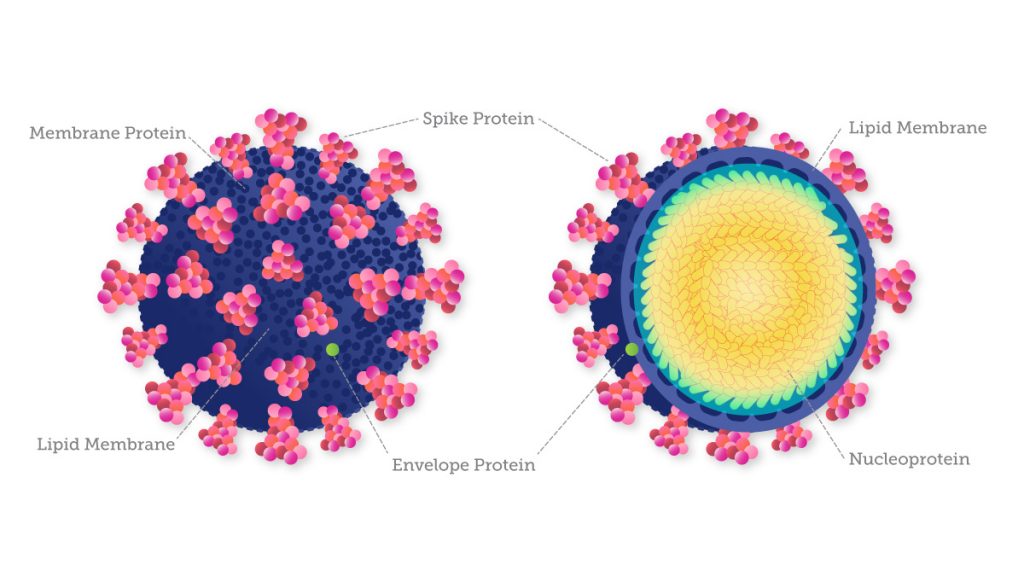
Like other coronaviruses, the genome of SARS-CoV-2 is composed of a single strand of RNA with a positive strand (ready for translation and consequent synthesis of its proteins). While its genome is comparatively large for a virus, it only has 29,903 base pairs (compared to the human genome’s more than three billion base pairs).
A method like shotgun metagenomics can be extremely useful in the case of a virus that is very contagious, is affecting large populations, and may be changing quickly. SARS-CoV-2, the virus responsible for the Covid-19 pandemic, was first identified in China in late December 2019. By January 12th, the Chinese authorities shared the full sequence of the coronavirus genome, as detected in samples taken from the first patients.
On the morning of January 24, the Institut Pasteur received samples of three suspected cases. The same evening, scientists confirmed the cases and began preparing the samples. The sequencing run was completed by early Tuesday evening, and the scientists used data analysis to obtain the sequence of the whole genome in two of the first three confirmed cases in France. They used the third sample to confirm their findings, and by Thursday, January 30, 2020, the Institut Pasteur was sharing the whole sequence of the virus.
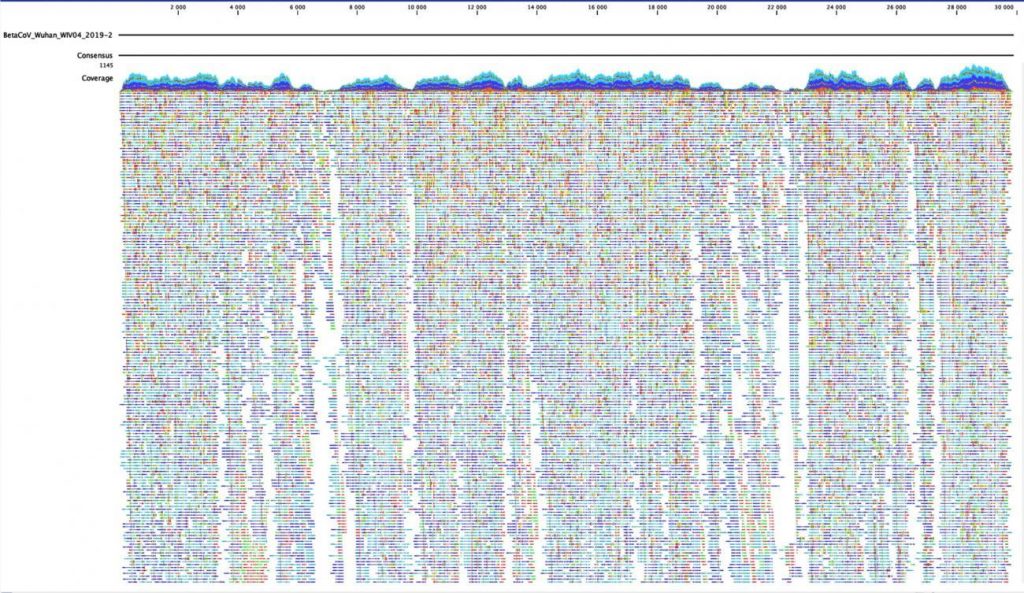
This kind of rapid sequencing and sharing, enabled by these kinds of next-generation methods, is extraordinary. By the end of April 2020, more than 12,000 SARS-CoV-2 genome sequences had been uploaded to GISAID, an open access database that tracks viral evolution and spread around the world. Still, this number lags far behind the number of cases, highlighting the difficulty in containing the spread of the virus and keeping track of its genetic sequence over time.
A Moving Target
Viruses like SARS-CoV-2 continuously evolve, as changes in their genome (mutations) occur during replication. More replications mean more potential mutations. More cases mean the virus can change very quickly in multiple ways, creating new variants. These mutations can confuse other methods that are looking for very specific sequences to identity a pathogen.
To inform local outbreak investigations and understand national trends, scientists need to compare genetic differences between viruses to identify variants and how they are related to each other. And to predict where they’re going.
Already, multiple variants of SARS-CoV-2 have been documented. The Delta variant was first detected in India in December 2020 and became the most commonly-reported variant in the country from mid-April 2021. It could be more than twice as transmissible as the original strain of SARS-CoV-2.

Knowledge of the virus’s genetic make-up also allows researchers to understand how SARS-CoV- 2 is evolving to monitor transmission and ensure that diagnostic tests and treatments that target viral genome products remain effective. For example, the current variants of concern show distinctive mutations in the spike protein, which help the virus infect healthy cells and even damage others. Because of these mutations, most diagnostic tests for COVID-19 are designed to target these spike proteins as well as other conserved proteins. Molecular tests designed to detect multiple SARS-CoV-2 genes are less susceptible to the effects of genetic variation than tests designed to detect a single gene.
What a deeper understanding of the disease looks like
This shotgun approach is crucial if novel pandemic pathogens like SARS-CoV-2 are to be tracked as they develop. Because we’re looking at everything (not just SARS-CoV-2) in a sample it can also provide us with information on patients’ responses to infection. A single nasal swab can provide a snapshot of the patient’s whole microbiome at a given time and place. We see not only viral RNA but also RNA from the patient’s own cells, helping us better understand how the immune system reacts to infection or how the virus can damage cells in ways we didn’t’ expect. We can detect coinfections and determine other organisms that may impact patient outcomes. This unlocks new discoveries related to why symptoms vary so much between patients, with some people almost totally unaffected and others in deadly danger.
There’s evidence that the microbiome of the respiratory tract can have an impact on the health of patients, meaning it could be possible to predict which patients with respiratory tract infections are more likely to experience more serious diseases by analyzing the microbiome.
Still, there are weaknesses. Because genomic sequencing is a complicated test that requires samples from individuals who have high viral loads, it is not easily used as a routine surveillance tool. Yet once it identifies worrisome variants in a region, routine laboratory tests can be created to track these variants in individuals with low viral loads who may not even know they have been infected, helping prevent further infections.
Since December 2020, the U.S. has sequenced only about 0.3% of individuals testing positive for Covid-19, with wide variation between states. The United Kingdom, in contrast, has consistently been sequencing 10% or more. The concern around low and inconsistent rates of sequencing is that this puts entire nations at risk of missing new variant mutations that could crop up at any time, in any part of the country, with potentially devastating effects.
Other researchers are getting creative with attempts to samples that they can sequence. In Marseille, in the south of France, researchers used Illumina NovaSeq equipment to monitor SARS-CoV-2 RNA in the city’s sewage system. They tested more traditional PCR screening as well as direct sampling with shotgun-style sequencing. They found the latter direct method made it possible to observe a distribution of the variants comparable to that revealed by genomic monitoring in patients with better accuracy than PCR. This could prove to be a powerful but relatively unintrusive tool for improving our understanding of the outbreak transmission dynamics and, also, of reducing the burden of COVID-19.
Other researchers found that metagenomic sequencing consistently helped detect pathogens that routine methods tended to miss – ideal if you’re looking for variants or coinfections you haven’t thought to look for. Yet others have looked at tracking infection breakouts between long-term care facilities.
Looking forward
At this point, the coronavirus disease global pandemic has left few completely unaffected. While no amount of science can make up for the losses we’ve all experienced, it can give us hope for the future – that future pandemics can be detected, contained, and treated faster and more effectively.
This pandemic has ignited the ingenuity of scientists, health care providers, and engineers to develop technologies that help us better:
- Identify vaccine targets for mRNA-based vaccines
- Track the transmission routes of the virus globally
- Detect mutations quickly to prevent the spread of new strain types
- Identify viral mutations that can avoid detection by established molecular diagnostic assays
- Identify viral mutations that can affect vaccine potency
- Screen targets for possible COVID-19 therapeutics
- Identify and characterize respiratory co-infections and antimicrobial resistance alleles
Today next-generation sequencing methods are primarily used in research; they have not been yet cleared for medical care. Imagine a future where smaller and more powerful sequencing tests could be something that pediatricians could take to the bedside. Politicians and public health experts could use the kind of information collected in Marseille at a much larger scale, which informing policy and helps to limit future outbreaks.
Even good information about a virus cannot tell us everything we want to know, which is how the virus will respond to different environments and segments of the global population.
 Genomics takes its place in the race against COVID-19
Genomics takes its place in the race against COVID-19  Advancing genome sequencing at the speed of light
Advancing genome sequencing at the speed of light 